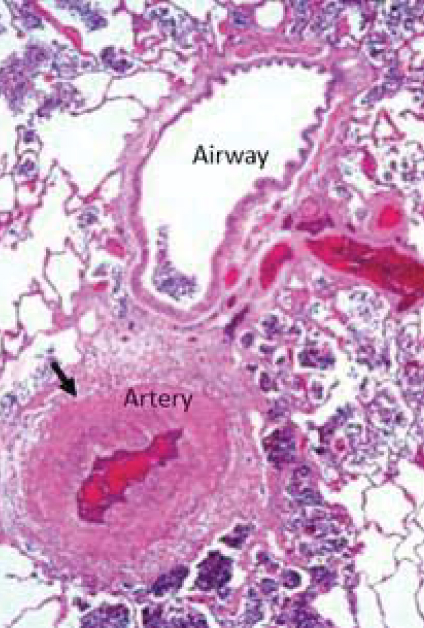
Pulmonary arterial hypertension (PAH) is a rare, progressive, life-threatening disorder characterized by increased pressure in the pulmonary arteries that carry blood from the heart to the lungs. The increased pulmonary artery pressure strains the heart, which can limit physical activity, cause breathlessness and fatigue during activities of daily living, and can ultimately result in heart failure and reduced life expectancy. PAH affects more than 30,000 patients in the U.S. Based on data from the Registry to EValuate Early And Long-term PAH disease management (REVEAL) of patients in the U.S., the estimated five-year survival rate of PAH following diagnosis is 57%.